Maligna oboljenja u dječijoj dobi
Broj djece oboljele od raka u svijetu se iz godine u godinu povećava. Međutim, veliki broj slučajeva ostaje nepoznat i neprijavljen zbog nepostojanja registra o broju djece oboljele od raka u velikom broju zemalja. U visoko razvijenim zemljama stopa preživljavanja doseže preko 80% i stalno se povećava. Maligne bolesti su drugi najčešći uzrok smrtnosti djece starije od 4 godine u razvijenim zemljama. Najčešće maligne bolesti kod djece su leukemije i limfomi, zatim tumori centralnog nervnog sistema i sarkomi. U dječijoj dobi vrlo česti tumori su neuroblastom, tumori bubrega, tumori jetre, tumori kostiju i retinoblastom.
Leukemije
Leukemije su maligne bolesti koje nastaju zbog nekontrolisane diobe krvnih stanica u koštanoj srži. Leukemične stanice potpuno infiltriraju koštanu srž, ulaze u perifernu krv i infiltriraju sva tkiva u tijelu. Uzrok akutnih leukemija nije poznat. Mogući faktori rizika su: virusi (HTLV-1 i HTLV-2, EBV), zračenje, hemijski spojevi (benzen), hromozomske promjene (npr. osobe s Down-sindromom, Klinefelterov sindrom), nasljedne bolesti (Fankonijeva anemija), imunodeficijentni bolesnici (npr. kod oboljelih od AIDS-a). Simptomi leukemija su: opća slabost, blijedilo, krvarenja po koži, iz nosa ili desni, povišena temperatura, bolovi u kostima i zglobovima.
Prije početka liječenja leukemije potrebno je obaviti detaljne labaratorijske pretrage koje uključuju:
● KKS i razmaze periferne krvi
● Aspiracijsku punkciju i biopsiju koštane srži
● Citohemijska bojenja ćelija periferne krvi i koštane srži
● Protočnu citometriju s imunofenotipskom analizom malignih ćelija
● Citogenetičku analizu malignih ćelija
● Molekularne testove
Akutna limfoblastna leukemija (ALL)
Predstavlja skupinu malignih bolesti kod kojih ćelije koje se normalno razvijaju u limfocite postaju maligne i u koštanoj srži zamjenjuju normalne stanice. Najčešća je maligna bolest kod djece. Odgovorna je za 25% svih malignih bolesti u dječijoj dobi. Uglavnom se javlja u periodu od druge do pete godine života.
KLINIČKA SLIKA
Simptomi i znaci zavise od stepena blastne infiltracije koštane srži i ekstramedularnih organa.
Prvi simptomi su:
● opća slabost (posljedica smanjenog broja eritrocita-anemija),
● blijedilo,
● krvarenja po koži, iz nosa ili desni (smanjen broj trombocita),
● infekcija i povišena temperatura (smanjen broj leukocita),
● bolovi u kostima i zglobovima.
Mogu se javiti simptomi i znaci od strane centralnog nervnog sistema (CNS) kao što je povećanje intrakranijalnog pritiska, glavobolje, konvulzije, poremećaji svijesti, vida i sluha, edem papile itd. Kod malog broja oboljelih mogu se javiti znaci cerebralne disfunkcije, halucinacije i poremećaji ponašanja.
DIJAGNOZA
Prvi dokaz da je dijete oboljelo od leukemije mogu biti krvne pretrage (KKS). Ukupan broj leukocita može biti snižen, normalan ili povišen, ali je broj eritrocita i trombocita uvijek snižen. U razmazu periferne krvi kod osoba oboljelih od leukemije, pod mikroskopom, vide se nezrele bijele stanice (blasti). Normalno blasti se ne vide u krvi, pa je njihovo prisustvo znak leukemije. Za potvrdu dijagnoze i određivanje vrste leukemije radi se biopsija koštane srži.
LIJEČENJE
Liječenje ALL se odvija u nekoliko faza. U liječenju se primjenjuju kortikosteroidi i citostatici. Prva faza je indukciona faza i njen cilj je postizanje remisije bolesti, što se manifestuje odsustvom blasta u koštanoj srži. Zatim slijedi primjena citostatika koji prolaze hemato-encefalnu barijeru da bi se uništili blasti u CNS-u, a nakon toga faza intenzivne citostatske terapije. Posljednja faza je podrazumijeva primjenu citostatika peroralnim putem. Cilj liječenja je uništavanje leukemičnih ćelija tako da u koštanoj srži mogu ponovo rasti normalne ćelije. Povoljan ishod se očekuje uglavnom kod tzv. uobičajene ALL porijeklom iz B limfocita, dok nepovoljniju prognozu ima akutna leukemija porijekla zrelih B limfocita. Najbolju prognozu imaju djeca u dobi između 3. i 7. godine života. Ponekad može doći do recidiva bolesti pa je kod takvih pacijenata najbolja metoda liječenja transplantcaija koštane srži. Preživljenje od akutne limfoblastične leukemije je u 75-80% slučajeva.
Akutna mijeloidna leukemija (AML)
Predstavlja maligno oboljenje kod kojeg mijelociti postaju maligni i brzo zamjenjuju normalne ćelije koštane srži. Faktori rizika za razvoj AML su: visoke doze zračenja i upotreba hemoterapijskih lijekova.
KLINIČKA SLIKA
Simptomi se javljaju kao posljedica disfunkcije koštane srži usljed infiltracije malignim ćelijama. Javlja se krvarenje uzrokovano diseminiranom intervaskularnom koagulacijom, te tumorske mase koje se nazivaju hloromi ili granulocitni sarkomi. Ove tumorske mase se javljaju u koži, orbiti i epiduralno.
DIJAGNOZA
Postavlja se na osnovu kompetne krvne slike (KKS), krvnih uzoraka ispitivanih pod mikroskopom, te biopsijom koštane srži. AML se od ALL može razlikovati pozitivnim citohemijskim bojenjem mijeloperoksidaze leukemijskih blasta.
LIJEČENJE
AML se liječi citostaticima, a vrlo često je potrebna i transplantacija matičnih ćelija koštane srži. Povoljan ishod se očekuje kod oko 60% oboljelih.
Hronična mijeloidna leukemija (HML)
Predstavlja bolest kod koje ćelije u koštanoj srži postanu maligne i pri tome proizvode veliki broj nenormalnih granulocita. Leukemični granulociti mogu nastati u koštanoj srži, jetri i slezeni. Ćelije se kreću od vrlo nezrelih do zrelih oblika. Imaju sklonost istjerivanja normalnih stanica koštane srži što dovodi do stvaranja velike količine vezivnog tkiva. Zbog toga je u krvnoj struji prisutan veliki broj nezrelih granulocita, te se razvija anemija i trombocitopenija, a povećava se i broj blasta. Uglavnom se javlja kod odraslih, a kod djece vrlo rijetko. Javlja se umor, slabost, gubitak apetita, visoka temperatura, noćno znojenje i osjećaj punoće. Također, može se javiti i visok broj leukocita, hepatomegalija i splenomegalija. Bolest je u 99% slučajeva obilježena pojavom translokacije t (9;22) koja se naziva Philadelphia hromosom.
Dijagnosticira se na osnvu krvnih pretraga (KKS, krvni razmaz), hromozomske analize.
Liječi se hemoterapijom, a najbolji ishod je transplnatacija koštane srži.
Tumori CNS-a
Tumori centralnog nervnog sistema (CNS) javljaju se kod djece svih uzrasta, a najčešće do 7. godine života. Kod djece većina tumora je primarna, a etiologija je najčešće nepoznata. To su najčešći solidni tumori kod djece, a u ukupnoj učestalosti malignih bolesti su na drugom mjestu, poslije akutnih leukemija. Glavni su uzrok smrtnosti pedijatrijskih onkoloških pacijenata.
ETIOLOGIJA
Javljaju kod bolesnika koji su liječeni radioterapijom i kod bolesnika sa urođenim sindromima: neurofibromatoza tip I i II, von Hippel-Lindau, tuberozna skleroza, Li-Fraumeni i Turcotov sindrom. Riziko faktori za razvoj tumora CNS-a su: prehrambene navike majke tokom trudnoće (namirnice koje sadrže nitrozamin), pušenje (oba roditelja). U tumore mozga ubrajaju se astrocitomi (čine 50% svih tumora CNS-a), tumori moždanog stabla (15-20% svih tumora mozga), ependimomi, gliomi, meduloblastomi i kraniofaringeomi.
KLINIČKA SLIKA
Tokom prve godine života tumori CNS-a se najčešće ispoljavaju povećanjem obima glave, kao prvim znakom povišenog intrakranijalnog pritiska. Znak uznapredovalog povećanja intrakranijalnog pritiska je Cushingov trijas, koji čine bradikardija, hipertenzija i tahipneja. Najčešći simptomi su: glavobolja, mučnina, povraćanje, epileptički napadi, poremećaji vida i ataksija. Kod mlađe djece javlja se razdražljivost, gubitak apetita, zaostajanje u razvoju i regresija usvojenih intelektualnih i motoričkih vještina. Kod školske djece simptomi su povezani sa slabijim uspjehom u školi i slabom koncentracijom. Javlja se umor i česte promjene raspoloženja. Cerebelarne tumore karakteriše poremećaj koordinacije, ataksija i povišen intrakranijalni pritisak. Tumore kičmene moždine karakteriše bol u leđima, poremećaj koordinacije i ravnoteže, deformitet kičmenog stuba i poremećaj funkcije sfinktera.
DIJAGNOZA
Dijagnostičke metode uključuju anamnezu, fizikalni pregled, neurološki pregled, CT, MR, PET, angiografiju i lumbalnu punkciju.
LIJEČENJE
Mogućnost preživljavanja kod djece s primarnim tumorom mozga je u 55-60% slučajeva. Hirurška terapija je neophodna za većinu tumora mozga. Ukoliko je moguće kao metoda izbora radi se kompletna resekcija. U liječenju većine tumora mozga, uz hiruršku, koristi se i radioterapija i sistemska hemoterapija.
Limfomi
Limfomi su maligni tumori limfnog tkiva. Dijele se na: Hodgkin limfom i non-Hodgkin limfom. Nakon leukemija i tumora CNS-a, limfomi su treće najčešće maligno oboljenje kod djece. Uglavnom obolijevaju djeca starija od pet godina i adolescenti.
Hodgkinov limfom (HL)
Hodgkinov limfom je maligna bolest koja nastaje u limfnim čvorovima. Predstavlja najučestaliju malignu bolest mlađe djece i adolescenata od 14. do 19. godine.
ETIOLOGIJA
Faktori rizika za razvoj Hodgkinovog limfoma su: infekcije (npr. Epstein-Barrov virus (EBV)), genetski poremećaji (HL se češće javlja kod blizanaca)
Na osnovu kliničkih i morfoloških karakteristika, te biološkog ponašanja i imunofenotipa Hodgkinov limfom je klasificiran na: nodularnu limfocitnu predominaciju Hodgkinovog limfoma (NLPHL) i klasični Hodgkinov sindrom (KHL).
KLINIČKA SLIKA
Uvećanje površnih limfnih čvorova je najčešći znak HL. Limfni čvorovi su bezbolni, čvrste konzistencije, palpiraju se pojedinačno ili u grupama. Većina oboljelih ima uvećane limfne čvorove u području vrata i pazušne regije. Vrlo često je zahvaćen medijastinum što uzrokuje nadražajni, neproduktivni kašalj i otežano disanje. Javlja se noćno znojenje, svrbež kože, gubitak tjelesne težine i blago povišena tjelesna temperature. Može biti uvećana slezena i jetra. U slučajevima kad je zahvaćena koštana srž javlja se anemija, leukopenija i trombocitopenija.
Na osnovu rasprostranjenosti bolesti HL je klasificiran u četiri klinička stadija:
● I stadij- limfni čvor u jednoj regiji ili pojedinačna ekstranodalna klasifikacija
● II stadij- limfni čvorovi dvije ili više nazavnisnih anatomskih lokalizacija, sa iste strane dijafragme,
● III stadij-limfni čvorovi sa obje strane dijafragme
● IV stadij-zahvaćenost jednog ili više ekstranodalnih organa ili tkiva, sa ili bez zahvaćenosti limfnih čvorova.
DIJAGNOZA
Postavlja se na osnovu patohistološkog nalaza uzorka tumora. Imunohistohemijske metode koriste se za klasifikaciju limfoma, dok aspiracija ili biopsija koštane srži, te pregled cerebrospinalnog likvora služe za procjenu rasprostranjenosti bolesti. U krvnoj slici može biti snižen broj ćelija jedne ili više krvnih loza kao posljedica infiltracije koštane srži. Ponekad mogu biti prisutni znaci hemolize, Coombs pozitivne anemije ili eozinofilija. Prisutna je sedimentacija eritrocita, a serumske koncentracije fibirnogena su povišene. Za preciznu procjenu proširenosti bolesti potrebno je uraditi CT grudnog koša i trbuha.
LIJEČENJE
Zadovoljavajući rezultati postižu se hemoterapijom (kombinacija kortikosteroida i citostatika). Terapija zračenjem se koristi samo za uznapredovale oblike HL.
Non-Hodgkinov limfom
Non-Hodgkinov limfom je maligni tumor koji nastaje u limfnom čvoru. Češće se javlja kod dječaka nego kod djevojčica. Vrlo je agresivan i brzo se širi u koštanu srž, te ga je ponekad teško razlikovati od akutne limfoblastne leukemije.
ETIOLOGIJA
Faktori rizika koji mogu uzrokovati nastanak non-Hodgkinovog limfoma su:
● virusi (HTLV-1, EBV)
● autoimmune bolesti
● urođena imunodeficijentna stanja
● sekundarne imunodeficijencije
● velike doze zračenja
Postoje tri osnovna oblika non-Hodgkinovog limfoma:
B-ćelijski (“Burkitt” i “non-Burkitt”-limfomi malih “zarezanih” ćelija, te B-velikoćelijski limfomi); Limfoblastični; Velikoćelijski anaplastični.
KLINIČKA SLIKA
Prvi uočljiv simptom je povećanje limfnog čvora u području vrata, prepona ili po cijelom tijelu. Povećani limfni čvorovi u krajnicima uzorkuju tečkoće pri gutanju, a u grudnom košu i trbuhu mogu uzrokovati otežano disanje, gubitak apetita, zatvor, bol u trbuhu ili oteknuće noge. Ako limfom dospije u krv može dovesti do razvoja leukemije. Kod djece može doći do anemije, osipa i slabosti.
DIJAGNOZA
Dijagnoza se postavlja na osnovu patohistološke analize limfnog čvora i imunofenotipizacije tkiva tumora. Za utvrđivanje proširenosti bolesti koristi se CT ili MR. Prisutna je ubrzana sedimentacije eritrocita i povećana aktivnosti laktat-dehidrogenaze (LDH). Ovisno o proširenosti može doći do anemije, povećanja vrijednosti mokraćne kiseline i alkalne fosfataze.
LIJEČENJE
U liječenju se najčešće koristi hemoterapija, uz primjenu kortikosteroida. U slučaju neuspjeha hemoterapije bolest se liječi transplantacijom koštane srži.
Neuroblastom
Neuroblastom je najčešći ekstrakranijalni solidni tumor dječijeg doba. Nastaje iz neuralnog grebena, a građen je od neoplastičnih neuroblasta. Najčešće primarne lokalizacije neuroblastoma su srž nadbubrežne žlijezde i paraspinalne ganglije. Ćelije neuroblastoma sintetišu i luče kateholamine (epinefrin, norepinefrin i dopamin) čiji metaboliti vanilmandelična kiselina (VMA) i homovanilična kiselina (HVA) mogu biti prisutni u urinu, serumu ili tumorskom tkivu. Zbog prognoze u tumorskom tkivu se radi pretraga broja kopija proonkogena N-myc, jer više od šest kopija upućuje na vrlo lošu prognozu.
KLINIČKA SLIKA
Zavisi od lokalizacije tumora. U dojenčadi i male djece tumor je uglavnom u trbušnim paravertebralnim simpatičkim stanicama ili u srži nadbubrežne žlijezde. Kada tumorska masa postane dovoljno velika da stvara pritisak na druge organe (crijeva, jetru, bubreg ili kičmenu moždinu) javljaju se prvi simptomi. Veliki abdominalni tumor može uzrokovati infiltraciju jetre i dispneju zbog potiskivanja dijafragme. Tumor u predjelu vrata često uzrokuje stridor, disfagiju i Hornerov sindrom (ptoza, mioza i enoftalmus). Ponekad se može javiti i blijedilo jedne polovine lica (znak harlekina). Tumorska masa u grudnom košu javlja se kod starije dijece i praćena je dispnejom. Metastaziraju u okolne limfne čvorove, jetru i pluća. Uzrokuje povećanje abdomena, hepatomegaliju, ascites i respiratorni distres sindrom. Usljed sekrecije vazoaktivnih intestinalnih peptida javljaju se teški proljevi. U mokraći su povećane količine norepinefrina, homovanilične kiseline (HVA) i dopamina. Djeca su febrilna, gube na težini, javljaju se bolovi u zglobovima, mogu imati paraneoplastične simptome poput ataksije ili opsomioklonusa (“nemirne oči i stopala”). Zbog arterijske hipertenzije ili nedostatka autoimunog odgovora organizma na tumor može se javiti encefalopatija. Poremećaji vida mogu nastati zbog metastaza u orbiti i infiltracije optičkog živca. Manje od 2% bolesnika ima paraneoplastične sindrome (sindrom opsoklonus-mioklonus-ataksija; sindrom vodene dijareje).
DIJAGNOZA
Preko 90% oboljele djece je mlađe od 5 godina. Zlatni standard u postavljanju dijagnoze neuroblastoma je ispitivanje tkiva primarnog tumora ili metastaza histopatološkim ili imunohistohemijskim metodama. Tumorsko tkivo služi za određivanje genetskih markera. Za procjenu stadijuma bolesti i određivanje grupe rizika neophodno je uz radiografske metode uraditi scintigrafiju skeleta, pregled koštane srži sa određivanjem kariotipa. Od laboratorijskih nalaza povišena je brzina sedimentacije eritrocita, koncentracija serumskog feritina, neuron-specifične enolaze i laktat-dehidrogenaze u serumu. U 24-satnom urinu povišene su koncentracije homovanilinske ili vanilmandelične kiseline. Tri dana prije početka prikupljanja uzorka urina treba provesti posebnu dijetu, zbog mogućeg lažno pozitivnog nalaza. Stanice neuroblastoma nakupljaju radioaktivni jod (MIBG= metajodobenzilgvanidin), što služi otkrivanju metastaza i kasnije za praćenje aktivnosti bolesti.
LIJEČENJE
U prvom i drugom kliničkom stadiju dovoljna je hirurška resekcija tumora, dok se u odmaklim stadijima primjenjuje hemio i radioterapija. Pri liječenju hemoterapijom koriste se cisplatin, karboplatin, doksorubicin i ciklofosfamid. U fazi održavanja remisije može se davati i cis-retinoična kiselina.
Tumori jetre
Tumori jetre su treći najučestaliji intraabdominalni tumor kod djece. U dječjoj dobi su dosta rijetki i čine svega 1-2% tumora u toj dobi. Mogu biti maligni i benigni. Maligni tumori se češće javljaju nego benigni. Naučestaliji maligni tumori jetre su hepatoblastom i hepatocelularni karcinomi. Hepatoblastomi se najčešće javljaju oko treće godine života, dok hepatocelularni karcinomi mogu da se jave i kod male djece, ali i kod adolescenata.
ETIOLOGIJA
Postoji veći broj riziko faktora koji mogu uzrokovati nastanak tumora. Uglavnom se dijele na faktore koji se odnose na majku djeteta (uzimanje oralnih kontraceptiva, duhan, alkohol), kao i faktori vezani za samo dijete (nasljedne, metaboličke i zarazne bolesti). Tumor može nastati i kao posljedica infekcije HBV i HCV, a kao veliki problem smatra se nedostatak vakcine protiv HCV, kao i neprovođenje imunizacije protiv HBV u siromašnijim krajevima. Nevirusne bolesti jetre, ciroza, tirosemija i bilijarna ciroza su povezane s nastankom hepatocelularnog tumora.
KLINIČKA SLIKA
Tumore jetre karakteriše pojava abdominalne mase ili distenzija abdomena. Najvažniji simptomi su: gubitak apetita i smanjenje tjelesne težine, proljevi ili zatvori, nemir i slab san, slabost i umor, te oticanje trbuha. Kasnije može doći do kašlja, visoke temperature, ascitesa, teške boli, ikterusa i mučnine.
DIJAGNOZA
Pregled svakog pacijenta započinje uzimanjem anamneze i fizikalnim pregledom. Zatim je neophodno uraditi laboratorijsku dijagnostiku, koja obuhvata:
⮚ kompletnu krvnu sliku
⮚ analizu urina
⮚ jetrene probe
⮚ elektrolite
⮚ faktore koagulacije (fibrinogen, protrombinsko vrijeme, parcijalno trombloplastinsko vijeme)
⮚ testiranje na viruse (HAV, HBV; HVC, CMV, EBV, HIV-1)
⮚ tumorski markeri (AFP, β-hCG, CEA)
Osim laboratorijskih pretraga, u dijagnozi tumora jetre koriste se i radiološke metode: ultrazvuk, kompjuterizovana tomografija (CT) i magnetna rezonanca (MR).
LIJEČENJE
Zahvaljujući hirurškoj terapiji, radioterapiji i hemoterapiji kao i transplantaciji jetre, preživljavanje djece s tumorima jetre se u posljednjih nekoliko desetljeća znatno povećalo.
Tumori bubrega
Nefroblastom ili Wilmsov tumor je embrionalni tumor porijekla od primitivnih embrionalnih ćelija bubrega i najčešći je primarni tumor bubrega kod djece. Većinom se javlja u dobi od 2-5 godina i uglavnom je lokalizovan na jednom bubregu, a u oko 5% djece se javlja obostrano i to ili istovremeno (sinhroni tumor) ili prvo na jednom, a potom na drugom bubregu (metahroni bubreg). Često je udružen s više kongenitalnih anomalija (aniridija, hemihipertrofija, anomalije urogenitalnog sistema), sindroma i hromozomskih aberacija. Anomalije povezane sa Wilmsovim tumorom povezane su sa gubitkom genetskog materijala na hromosomu 11p na kojem se nalazi i tumorosupresorski gen WT1.
KLINIČKA SLIKA
Pacijenti se najčešće jave zbog tegoba koje nastaju zbog veličine tumora. Prisutna je palpabilna abdominalna masa koja se širi preko središnje linije i prema zdjelici. Rjeđe se prvo javlja visoka temperatura i bol u trbuhu sa hematurijom ili crijevna opstrukcija zbog pritiska tumora, te hipertenzija. Kod malog broja djece zbog krvarenja u tumoru mogu postojati znaci anemije.
DIJAGNOZA
Postavlja se UZ pregledom i drugim radiološkim dijagnostičkim postupcima. Istovremeno uz patohistološki pregled određuje se tip tumora. U većini slučajeva Wilmsovog tumora radi se o histološkim oblicima sa povoljnom prognozom. Stadij bolesti se utvrđuje da bi se odabralo prikladno liječenje. Prije operativnog odstranjenja tumora potrebno je napraviti i infuzijsku urografiju da se dokaže normalna funkcija zdravog bubrega. Rendgen pluća je dovoljan za otkrivanje metastaza u plućima. Kod nefroblastoma se u plućima mogu naći lažno pozitivni čvorići, koji se sastoje od agregata limfocita, fokalnih atelektaza ili granuloma.
LIJEČENJE
Provodi se citostatska terapija (bez prethodne biopsije) u cilju smanjenja tumora. Prognoza je općenito dobra i odlični se rezultati postižu kombinacijom radioterapije, nefrektomije i hemoterapije. Djeca s tumorima povoljne histologije i u prva tri stadija bolesti u 85-98% slučajeva se oporave od bolesti, dok oni s nepovoljnom histologijom preživljavaju u oko 60% slučajeva. Veliki problem su bilateralni tumori, kada se operativnim zahvatom odstranjuje bubreg koji je više zahvaćen tumorom, a na drugoj strani se uradi parcijalna nefrektomija.
Tumori kostiju
Rabdomiosarkom
Rabdomiosarkom je najčešći mekotkivni sarkom kod djece s incidencom od 5-6 novootkrivenih bolesnika na milijun djece godišnje. Najčešće primarno mjesto tumora su glava i vrat (40%), mala zdjelica (20%), ekstremiteti (20%) i trup (10%). Nastaje od mezenhimnih ćelija odgovornih za nastanak poprečno prugastih mišića.
KLINIČKA SLIKA
Klinički se rabdomiosarkom očituje kao bezbolna oteklina. Simptomi se javljaju tek kada pritisne okolne anatomske strukture. U ždrijelu može izazvati otežano disanje i gutanje, a ponekad i epistaksu. U uhu uzrokuje slabljenje sluha, a bol se javlja kada prodire u kost. U gingivi može izgledati kao upalni proces. Može rasti prema bazi mozga, te izazvati glavobolju i parezu kranijalnih živaca. U orbiti može izazvati protuziju bulbusa. Na ekstremitetima podsjeća na hematom. U urogenitalnom sustavu uzrokuje hematuriju. Metastazira u okolne limfne čvorove i pluća.
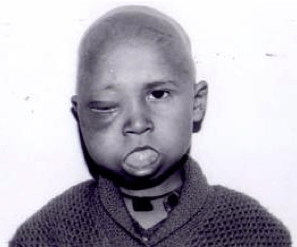
Slika 4. Rabdomiosarkom
DIJAGNOZA
Postavlja se patohistološkim pregledom uzorka tumorskog tkiva, ali za određivanje podtipova neophodna je imunohistohemijska i citogenetska analiza kojom se nalaze specifične translokacije hromozoma. U diferencijalnoj dijagnozi važna je lokalizacija.
LIJEČENJE
Liječenje ovisi o proširenosti, mogućnosti hirurškog odstranjenja tumora u cijelosti i histološkim karakteristikama tumora. U većini slučajeva nakon postavljanja dijagnoze provodi se preoperativna hemoterapija, a nakon toga se nastoji u cijelosti odstraniti tumor. Ako se tumor u potpunosti može odstrasniti i ukoliko je histologija povoljna izlječenje je moguće u 70-80% slučajeva.
Osteosarkom
Osteosarkom je maligni mezenhimalni tumor građen od neoplastičnih osteoblasta koji aktivno stvaraju osteoid ili kalcifikovanu kost. Najčešće se javljuju u starosnoj dobi od 10-20 godina s incidencom od oko 6-7 oboljelih na milion djece do 16 godina života. Muškarci oboljevaju nešto češće nego žene u odnosu 6:1.
Postoji nekoliko podtipova osteosarkoma:
• Primarni • Solitarni • Intermedularni • Slabo diferencirani
Uzroci osteosarkoma nisu poznati, ali se smatra da genetski faktori imaju značajnu ulogu u patogenezi. Mutacije gena RB-1 nalaze se u oko 65% svih osteosarkoma, a mutacije p53 i cijelog niza onkogena mogu biti prisutne kod većine osteosarkoma.
KLINIČKA SLIKA
Očituje se oteklinom i boli u dugim kostima. Javlja se bolna, rastuća masa koja onemogućava normalnu pokretljivost. Vrlo često djeca navode traumu kao uzrok nastanka otoka. Iz tog razloga svaki otok na kostima koji ne prolazi u očekivanom vremenskom periodu treba dijagnostički rasvijetliti. Ponekad se oko otoka javlja upalna reakcija okolnog mekog tkiva. Bol u kostima se javlja noću i remeti djetetu san. Rendgenski snimak pokazuje veliku tumorsku tvorevinu nejasnih rubova. Ako se tumor širi u okolna meka tkiva na granici tumora i probijenog korteksa nalazi se karakterističan trougao koji predstavlja odignuti periost i to se naziva Codman-ovim trouglom. Prisutni su i zrakasti koštani tračci poput izlazećeg sunca.
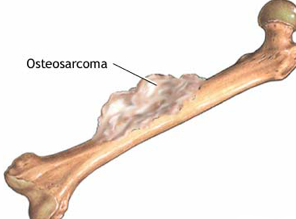
Slika 5. Osteosarkom
DIJAGNOZA
Dijagnoza se postavlja hirurškom biopsijom kosti. Da bi se procijenila proširenost bolesti potrebno je uraditi scintigrafiju. Osteosarkom metastazira u okolna tkiva i pluća.
LIJEČENJE
Liječenje započinje hemoterapijom. Nakon smanjenja tumorske mase hirurški se odstranjuje tumor radi očuvanja ekstremiteta. Tokom odstranjenja tumora ili kasnije može se ugraditi i proteza. Potom slijedi dovršetak hemoterapije. Stopa preživljavanja je u 65-75% slučajeva, a lošiju prognozu imaju bolesnici s plućnim metastazama, te oni kod kojih su zahvaćene kosti trupa.
Ewingov sarkom (Juingov sarkom)
Ewingov sarkom, odnosno “skupinu Ewingovih sarkoma” čine Ewingov sarkom kosti, Ewingov sarkom mekih tkiva, te periferni primitivni neuroektodermalni tumor (PPNET). To je maligni tumor građen od nediferenciranih ćelija za koje se smatra da su matične, neuroektodermalne. Ima iste biološke osobine kao i primitivni neuroektodermalni tumor (PNET) mekih tkiva. Nakon osteosarkoma i hondrosarkoma treći je po učestalosti primitivni maligni tumor kosti. Najčešće se javlja u dobi od 5-20 godina.
Mehanizam nastanka tumora nije u potpunosti razjašnjen. Smatra se da svi ovi tumori nastaju iz neuroektodermalnih ćelija koje tokom fetalnog razvoja 'zalutaju' u kosti i meka tkiva. Kod većine Ewingovih sarkoma i PNET prisutna je hromosomska translokacija T(11;22)(q24;q12) koja prouzrokuje fuziju EWS gena s genom za transkripcijski faktor FL-1. Ovaj fuzionisani gen EWS-FL-1 funkcioniše kao onkogen koji stimuliše rast tumorskih ćelija. Kod preostalih 15% tumora koji ne pokazuju ovu translokaciju, javljaju se translokacije koje dovode do stvaranja onkogena koji potiču rast tumorskih ćelija.
KLINIČKA SLIKA
Kod polovine bolesnika javlja se bol u kostima. Zahvaćeno područje je otečeno, toplo, te bolno na dodir. Djeca često navode neku traumu kao početak simptoma. Otok kosti može biti praćen povišenjem tjelesne temperature, gubitkom na težini i jakim otokom okolnih mekih tkiva. Klinička slika oponaša infekciju, javlja se groznica, leukocitoza, anemija i povišena sedimentacija.
Slika 6. Ewingov sarkom
DIJAGNOZA
Radiološki se prikazuju kao litične promjene u dijafizi, koje su nejasnih rubova i povezane su sa periostalnom reakcijom u obliku novostvorenih koštanih lamela. Dijagnoza se postavlja temeljem patohistološke analize. Na RTG-u Ewingov tumor kosti nalikuje “lukovici”.
LIJEČENJE
U liječenju se kombinuju hirurška intervencija i hemoterapija. Mali tumori na ekstremitetima imaju dobru prognozu i stopu preživljavanja od oko 75%, dok tumori smješteni na trupu imaju dosta lošu prognozu s vrlo niskom stopom preživljavanja. Kod ove skupine tumora znaju se pojaviti kasni relapsi i 10-ak godina po prestanku liječenja.
Retinoblastom
Retinoblastom se javlja na troje od milion djece godišnje. U oko 60% oboljelih tumor je jednostran s negativnom anamnezom u porodici. Hereditarni oblik tumora može biti jednostran ili obostran. Na dugom kraku 13. hromosoma smješten je gen (RB1) koji kodira protein za koji se vjeruje da ima važnu ulogu pri nastanku retinoblastoma jer potiskuje tumorski rast.
KLINIČKA SLIKA I DIJAGNOZA
Dijagnoza se u 10% slučajeva postavlja rutinskim pregledom djece rođene u porodicama u kojima je prethodno bilo oboljelih od retinoblastoma. Ostali bolesnici se prepoznaju nakon rođenja ili u dojenačkoj dobi po bijelom pupilarnom odsjaju (tj. zjenica im je bijela). Osim bijelog pupilarnog odsjaja (leukokorija) može se pojaviti i strabizam, a u uznapredovalom obliku bolesti može se vidjeti upala tkiva orbite. Dijagnoza se postavlja oftalmološkim pregledom.
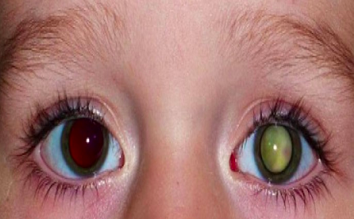
Slika 7. Retinoblastom
LIJEČENJE
Liječenje ovisi o veličini tumora. Jednostrani tumori liječe se enukleacijom bulbusa, što je u preko 95% slučajeva dovoljno za izlječenje. Obostrani tumori obično se liječe enukleacijom više zahvaćenog bulbusa, a prije odluke o enukleaciji drugog oka pokušava se liječiti zračenjem, krioterapijom ili fotokoagulacijom. Ako je tumor obostrano uznapredovao ili se proširio na okolna tkiva daje se hemoterapija, ali je prognoza za proširenu bolest loša. Djeca s pozitivnom mutacijom gena RB1 imaju veći rizik razvoja druge maligne bolesti, posebice osteosarkoma.
LITERATURA
- Damjanov I., Patologija, 5. izd., Medicinska naklada, Zagreb 2018. Godine
- Konja J, Aničić M (2009), Tumori jetre, Anđa Raič (ur.), Pedijatrijska onkologija, Zagreb, Medicinska naklada
- Mardešić D., Pedijatrija, 5. Izd., Medicinska naklada, Zagreb 2018. Godine
- MSD priručnici
- https://iccd.care/en-us/home
- https://www.ijzcg.me/me/medunarodni-dan-djece-oboljele-od-malignih-bolesti
- http://www.pedsoncologyeducation.com/braintumor_nomenclature.asp
- https://www.zzjzfbih.ba/medunarodni-dan-djece-oboljele-od-raka/
- https://zjztk.ba/view-more/medunarodni-dan-djece-oboljele-od-malignih-bolesti-2020/185