AMIOTROFIČNA LATERALNA SKLEROZA (ALS)
Amiotrofična lateralna skleroza, poznata kao ALS, rijetka je neurološka bolest koja pogađa motorne neurone u mozgu i kičmenoj moždini. Kako motorni neuroni odumiru, prestaju slati impulse mišićima, usljed čega dolazi do slabljenja mišića, fascikulacija i atrofije. Na kraju, mozak gubi sposobnost kontrole voljnih pokreta.
ALS je identificirao francuski neurolog Jean-Martin Charcot 1869. godine, ali je međunarodno poznat postao 1939. godine kada je završio karijeru igrača bejzbola, Loua Gehriga, po kome još i nosi naziv (Lou Gehrigova bolest). Bolest može biti sporadična (SALS) u kojoj ne postoji porodična historija bolesti i iznosi 90% slučajeva i porodična (FALS) koja se javlja u 10% slučajeva.
PATOFIZIOLOGIJA
Patologiju ALS-a karakterizira gubitak piramidalnih Betz ćelija u motornom korteksu, kao i gubitak i degeneracija velikih ćelija prednjih rogova kičmene moždine i donjih kranijalnih motornih jezgara moždanog stabla. Degenerirajući motorni neuroni pokazuju intracelularne agregate koji formiraju različita inkluzijska tijela: ubikvitinirane inkluzije (nalaze se uglavnom u donjim motornim neuronima moždanog stabla i kičmene moždine), Bunina tijela (male eozinofilne, hijalinske intracitoplazmatske inkluzije koje se pozitivno boje na cistatin C i transferin i smatraju se specifičnim za ALS) i inkluzije hijalinskog konglomerata (izrazito imunoreaktivni na neurofilamente i uglavnom se nalaze u motornim neuronima porodičnih ALS pacijenata).
Otprilike 25-40% svih porodičnih slučajeva (i mali postotak sporadičnih) uzrokovano je defektom gena C9ORF72, koji čini protein koji se nalazi u motornim neuronima i nervnim stanicama u mozgu. Neke osobe sa ovim genom također razvijaju tip frontotemporalne degeneracije (FTD, oblik demencije) uzrokovanu atrofijom temporalnog i frontalnog režnja mozga.
Patološke karakteristike ALS-a uključuju i agregaciju nerastvorljivih proteina unutar ćelija, s tim da tip proteinskog agregata varira među pacijentima. Smatra se da većina pacijenata ima akumulaciju TAR DNK vežućeg proteina 43 (TDP-43), dok mala grupa pacijenata ima akumulaciju superoksid dismutaze 1 (SOD1). Opisani su brojni mogući putevi bolesti, uključujući mitohondrijalnu disfunkciju, ekscitotoksičnost glutamate, probleme s autofagijom i izmijenjen metabolizam RNK. Također, smrt motornih neurona može biti “nećelijska autonomna”, što znači da druge vrste ćelija kao što su astrociti, mikroglije i eventualno oligodendrociti mogu uzrokovati smrt motornih neurona. Ranije se smatralo da je smrt neuronskih ćelija u ALS-u posljedica apoptoze koja je posredovana kaspazama. Dokazi za apoptozu kod ALS-a pronađeni su TUNEL bojenjem ljudskih tkiva i mjerenjem bcl-2 (porodica proteina odgovorna za kontrolu apoptoze). Drugi su otkrili povećan p53 kod ALS-a. Još jedan uobičajen oblik ćelijske smrti kod neurodegenerativnih bolesti je i nekroptoza, upalni oblik ćelijske smrti koji je nezavisan od kaspaze i uključuje aktivaciju RIP kinaze.
Bolest je progresivna, što znači da se simptomi vremenom pogoršavaju.
Rani simptomi uključuju:
· Trzanje mišića u nogama, rukama, ramenima i jeziku
· Grčenje mišića
· Ukočenost mišića
· Slabost mišića koja zahvata ruke, noge, vrat i dijafragmu
· Nerazumljiv ili nazalan govor
· Poteškoće u žvakanju i gutanju
Napredovanjem bolesti osobe gube mogućnost stajanja, hodanja, korištenja ruku i šaka, javljaju se disfagija, dizartrija, neuropatija, dispneja, koja se pogoršava vremenom, pa stanje zahtjeva korištenje mehaničkog ventilatora. Pacijenti također razvijaju anksiozni poremećaj i depresiju. ALS ne utiče na sposobnost okusa, dodira, mirisa ili slušanja. Većina ljudi s ALS-om umire od respiratorne insuficijencije, obično u roku od tri do pet godina od pojave prvih simptoma. Međutim, oko 10% ljudi s ALS-om preživi deceniju ili više.
Faktori rizika za ALS uključuju:
· Starost-Iako se bolest može pojaviti u bilo kojoj dobi, simptomi se najčešće razvijaju između 55. i 75. godine.
· Spol—muškarci imaju malo veću vjerovatnoću da razviju ALS. Međutim, starenjem razlika između spolova nestaje.
· Rasa i etnička pripadnost
DIJAGNOZA
Amiotrofičnu lateralnu sklerozu teško je rano dijagnosticirati jer može imati simptome slične drugim bolestima. Testovi za isključivanje drugih stanja ili pomoć u dijagnostici ALS-a mogu uključivati elektromiografiju (EMG), magnetnu rezonancu (MRI), nalaze krvi i urina, biopsiju mišića i nerava.
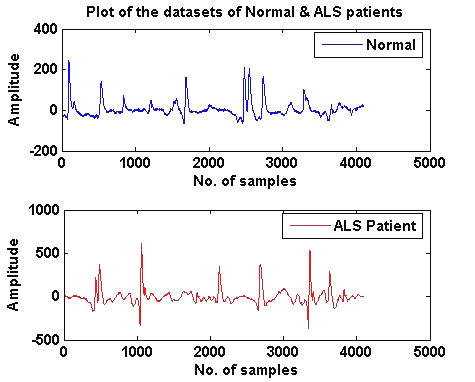
Slika 1. EMG zdravog i pacijenta sa ALS-om
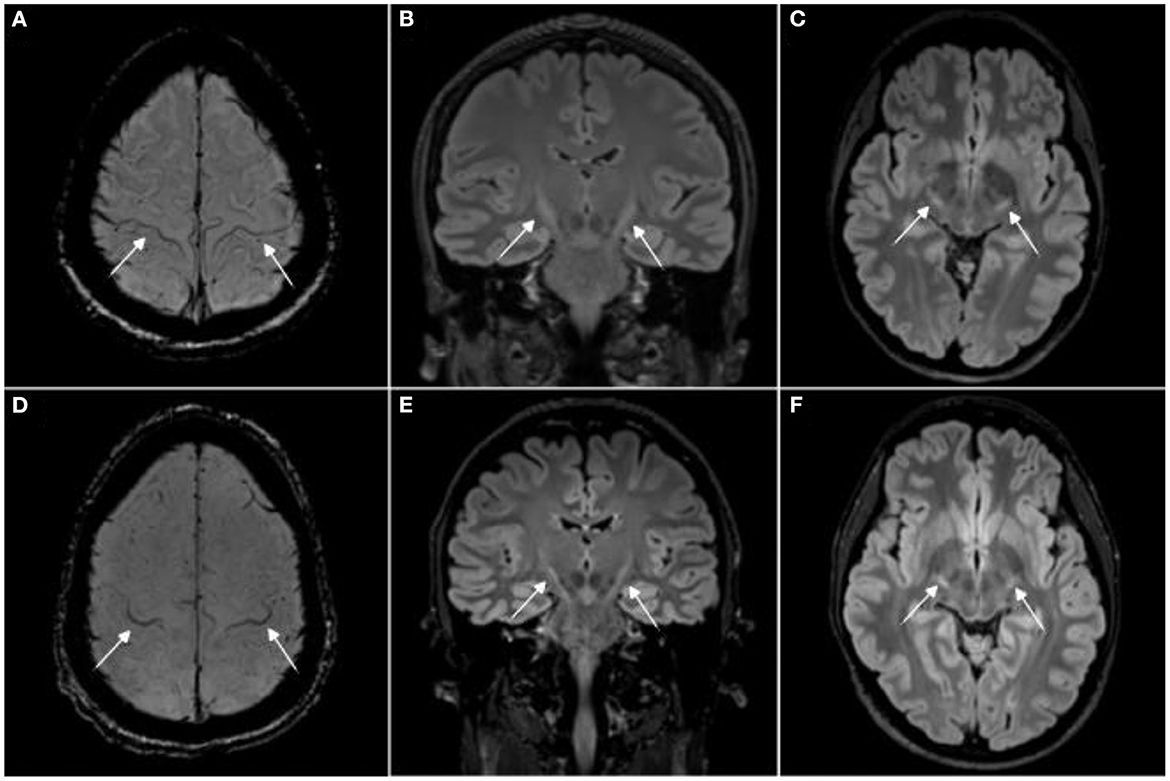
Slika 2. MRI pacijenta sa ALS-om
LITERATURA
1. Amyotrophic lateral sclerosis (ALS) (no date) National Institute of Neurological Disorders and Stroke. Available at:
https://www.ninds.nih.gov/health-information/disorders/amyotrophic-lateral-sclerosis-als
2. Understanding als (2023) The ALS Association. Available at:
https://www.als.org/understanding-als
3. Pathology of amyotrophic lateral sclerosis (no date) Pathology of Amyotrophic Lateral Sclerosis - an overview ScienceDirect Topics. Available at:
4. PMC - National Center for Biotechnology Information. Available at:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7186478/
5. Amyotrophic lateral sclerosis (ALS) (2023) Mayo Clinic. Available at:
6. EMG data pattern of a normal person and the ALS patient. Available at:
7. Cannon, A.E. et al. (2023) Neuroimaging findings in preclinical amyotrophic lateral sclerosis models-how well do they mimic the clinical phenotype? A systematic review, Frontiers. Available at:
https://www.frontiersin.org/articles/10.3389/fvets.2023.1135282/full