KONGENITALNA ADRENALNA HIPERPLAZIJA
Poznavajući građu nadbubrežne žlijezde, dalje se možemo osvrnuti na to šta se dešava u KAH!
Etiologija
KAH najčešće nastaje kao rezultat mutacije ili delecije CYP21A, ovaj gen je lokalizovan na hromozomu 6, a CYP21A kodira enzim 21ɑ - hidroksilazu. Preko 90% slučajeva KAH nastaje upravo zbog mutacije gena koji kodira enzim 21ɑ - hidroksilaza,a on je neophodan za biosintezu kortizola i aldosterona u organizmu. Težina bolesti zavisi od težine mutacije samog gena, a do sad je identificirano skoro 300 mutacija CYP21A. Također do KAH može doći i zbog nedostatka 11β-hidroksilaze, te zbog nedostatka 17ɑ-hidroksilaze, međutim ova dva nedostatka nisu toliko učestala.
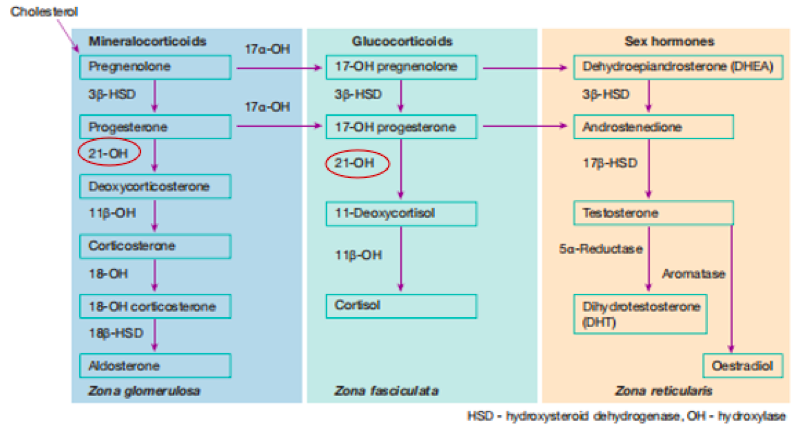
Svi hormoni nadbubrežne žlijezde imaju još jednu stvar zajedničku, a to je da je njihov prekursor holesterol, i da su svi hormoni steroidne građe, a u njihovoj biosintezi učestvuje niz enzima. Holesterol je hidrofobna supstanca, a u organizmu može biti dvojakog porijekla: egzogenog (iz hrane) i endogenog (sintetiziran de novo u organizmu). Za sintezu steroidnih hormona je ključan ovaj egzogeni holesterol koji se apsorbuje u enterocitima (ćelije tankog crijeva) i putem krvi, vezan za lipoproteine. U sintezi steroidnih hormona sudjeluju enzimi citohroma P450, ovi enzimi, monooksigenaze, učestvuju u prenosu elektrona sa NADPH na molekularni kisik preko proteina koji prenose elektrone.
U slučaju kongenitalne adrenalne hiperplazije, zbog mutacije gena koji kodira 21ɑ- hidroksilazu, a koja katalizira prevođenje progesterona u 11-deoksikortikosteron i 17ɑ-hidroksiprogesterona u 11-deoksikortizol dolazi do nemogućnosti sinteze aldesterona i kortizola. Kako je u organizmu smanjena produkcija kortizola i aldosterona, u hipofizi dolazi do pojačanog lučenja adenokortikotropnog hormona (ACTH) dovodeći do hiperplazije kore nadbubrežne žlijezde, što posljedično dovodi do akumulacije prekursora ovih hormona, progesterona i 17ɑ-hidroksiprogesterona. Nakupljanjem prekursora njihov metabolički put se preusmjerava u sintezu androgena, dihidroepiandosterona (DHEA) i androstenediona.
Patogeneza
Klinička slika ovog oboljenja je široka, a to je upravo zato što imamo različit nivo mutacije gena. Prekomjerna sinteza androgena može uticati na fetus, izazivajući ženski pseudohermafroditizam koji se kod ženskog novorođenčeta primjećuje, također kod ženske djece se može prije ili nakon puberteta javiti i virilizacija, to jeste, razvoj muških spolnih obilježja. Kod muške djece dolazi do prijevremeniog pseudopuberteta. Kod težih poremećaja može nastupiti i insuficijenicja nadbubrežne žlijezde, što posljedično dovodi do manjka aldosterona, te poremećajem prometa vode i elektrolita i kod takvih osoba postoji sklonosti ka nastanku cirkulatornog šoka. U slučaju manjih poremećaja enzima 21ɑ- hidroksilaze, koncentracija kortizola može biti i djelimično normalna, s tim da zalihe ovog hormona ne postoje.
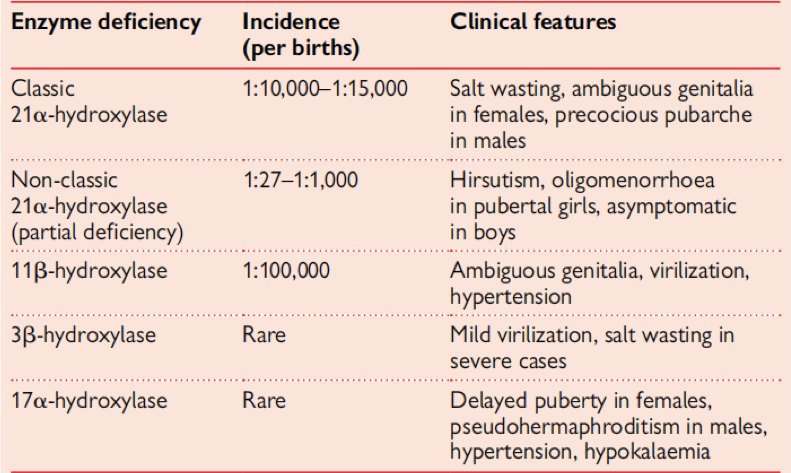
Postoje dvije forme KAH sa poremećajem 21ɑ-hidroksilaze:
1) Klasična kongenitalna adrenalna hiperplazija - većina pacijenata sa ovakvom formom bivaju identifikovani odmah po rođenju. U odrasloj dobi dolazi do niza problema. Kod žena dolazi do seksualne disfunkcije i subfertiliteta, još i po rođenju zbog virilizacije su potrebne rekonstruktivne genitalne operacije. Uz adekvatan medicinski tretman moguće je iznijeti normalnu trudnoću. Kod muškaraca zbog visokih nivoa adrenalnih androgena, dolazi do smanjenog lučenja gonadotropina, a time se smanjuje funkcija testisa, što dovodi do nastanka TART (engl.testicular adrenal rest tissue). Iako su uvijek benigni, mogu biti pogrešno dijagnosticirani kao maligni tumori testisa. Također, dovode do prestanka rada testisa. U neliječenim slučajevima KAH spermatogeneza je izuzetno niska.
2) Ne-klasična kongenitalna adrenalna hiperplazija - u kojoj imamo djelimičan nedostatak 21ɑ- hidroksilaze, gdje su nivoi aldosterona i kortizola zadovoljavajući, normalni, međutim dolazi do pretjerane produkcije androgena.U ovoj formi kod žena imamo kliničke simptome poput hirzutizma, akni, oligomenorejem, a mali broj pacijentica ima probleme sa začećem, dok muškarci prolaze asimptomatski.
Kod oboljelih osoba se zbog nepravilnog prometa elektroilta javlja povraćanje, dehidratacija, hipovolemija, što dalje može dovesti do šoka te u ekstremnim slučajevima smrti.
Fertilitet žena sa klasičnim oblikom KAH je smanjen, a razlozi su: poteškoće u spolnim odnosima zbog loših korekcija vagine i klitoromegalije, nezadovoljstvo izgledom i aseksualnost, anovulacijski cikusi itd. Kod muškaraca je također feritilnost smanjena, zbog TART-a te hipersekrecije adrenalnih adrogena.
Epidemiologija
U određenim populacijama pojava kongenitalne adrenalne hiperplazije je nešto češća nego u drugim populacijama. Tako na primjer, u Ujedinjenim Američkim državama, domorodci i yupik eskimi imaju nešto češću incidencu oboljenja, dok je kod ostale populacije prisutna u odnosu 1 na 15,000. Također imamo razliku u pojavi klasičnog i ne-klasičnog KAH u određenim populacijama, tako se ne-klasični češće javlja kod Aškenaza (19%), dok se klasični KAH javlja u odnosu 1:60 - 1:100 kod kavkasko naroda.
Dijagnostika
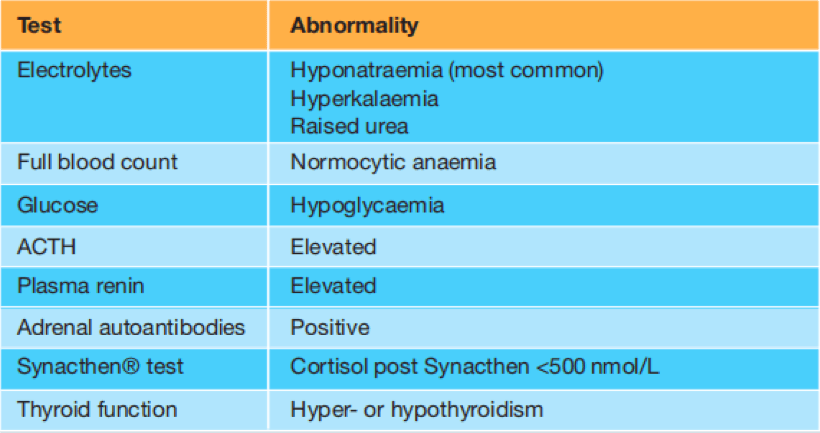
U dijagnostici KAH radi se screening test kojim se mjeri nivo 17ɑ-hidroksiprogesterona da bi se vidjelo da li postoji nedostatak 21ɑ.hidroksilaze. U slučaju pozitivnog testa, u ponovljenom testu će se leveli povišeno 17ɑ-hidroksiprogesterona isto održati, a pored ovog treba da se odredi i nivo elektrolita u serumu (Na, K...).
U slučaju klasičnog KAH nivoi 17ɑ-hidroksiprogesterona su veći od 242 nmol/L. Kod osoba koje imaju ovu autosomno recesivnu bolest zbog nedostatka aldosterona će imati hiponatrijemiju i hiperkalijemiju, a zbog nedostatka kortizola će nivo glukoze u krvi bit smanjen (hipoglikemija). Kod pacijenata sa dvosmislenim genitalijama, nekad je potrebno uraditi pelvični ultrazvuk - UZ za procjenu ostalih anomalija, te da bi se mogle definisati postojeće anomalije urogenitalnog trakta.
Kod djece sa preuranjenim pubertetom se može uraditi skeniranje kostiju radi pravilnijeg razvoja. Kod muškaraca je moguće otkriti TART ultrazvukom ili magnetnom rezonancom, a u rijetkim slučajevima palpacijom. Također postoje i drugi testovi koji se mogu raditi, kao naprimjer hromozomska analiza (radi se kod novorođenčadi sa dvosmislenim genitalijama radi određivanja spola), zatim genetičko testiranje u toku trudnoće, u nekim centrima se radi screening novorođenčadi na nedostatak 21ɑ-hidroksilaze da bi se spriječilo rasipanje soli koje može dovesti do letalnih posljedica.
Danas je moguća i dijagnostika fetalne DNK iz majčine krvi. Na taj način se može uraditi genotipizacija i odrediti već prije 5. sedmice da li fetus ima KAH, i u slučaju da se dokaže da ima, uvodi se terapija deksametazona što će omogućiti pravilan razvoj genitalija djeteta.
Liječenje i prevencija
U tretmanu ove bolesti važno je kontrolisati nivoe reproduktivnih hormona i njihov disbalans. Od postavljanja dijagnoze KAH, osobe moraju redovno koristiti glukokortikoidnu terapiju, mogu se koristiti hidrokortizon ili prednizolon. Da se ne bi terapijom razvila neka druga stanja, jako bitno je davati najniže terapijske doze ovih glukokortikoidnih lijekova. U najranijem dobu života, potrebe za glukokortikoidnom terapijom mogu biti nešto više, a u periodima stresa moguće je povećavati doze.
Kod dojenčadi sa klasičnim KAH sindromom također je potrebno koristiti i mineralokortikoidnu terapiju da bi se spriječio gubitak soli i posljedični poremećaji elektrolita. Tako da se daju fluorohidrokortizon acetat i NaCl (uz obrok). Nakon dojenačke dobi davanje NaCl-a više nije neophodno, a uglavnom se reducira davanje fluorohidrokortizona. Kod pacijenata sa jednostavnijim oblicima bolesti uglavnom postoje dovoljne količine aldosterona, te tretman mineralokortikoidima nije potreban.
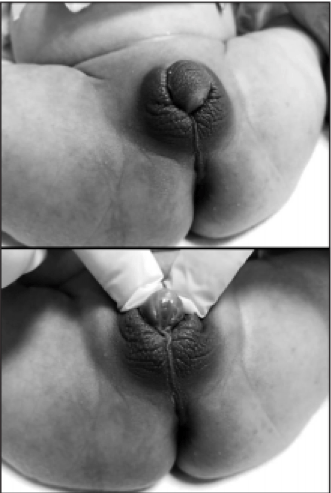
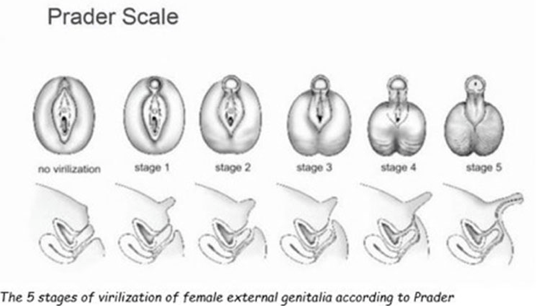
Kod pacijentica sa klasičniom kongenitalnom adrenalnom hiperplazijom je preporučena estetska i restruktivna hirurška intervencija, također preporuke se da se svi deformiteti pokušaju ispraviti u jednom aktu, i to prije prve godine života. Međutim, podijeljene su strane da li se treba sačekati da pacijentice sa dvosmislenim spolnim organima trebaju same odlučiti da li žele operaciju, a to bi trebalo biti nakon punoljetstva, i jako bitno je dobro genetski odrediti o kojem spolu se tačno radi. Jako je bitna psihološka terapija i pacijentica, a i njihovih roditelja i u periodu adolescencije je bitno smanjiti pregleda gentialija na minimum.
Trudnoća i KAH
Iako je feritilnost smanjena, trudnoća je kod liječenih oblika KAH moguća. U takvoj trudnoći je bitno voditi računa o tretmanu i o skrining testovima. Koristi se glukokortikoidna terapija i nekad se doze lijeka moraju udvostručiti. U liječenju trudnica je bitno ne koristiti deksametazon koji prolazi placentnu barijeru i na taj način može omesti razvoj fetusa, međutim u slučaju da se dokaže da fetus ima nedostatak enzima onda se deksametazon koristi već u trudnoći kao tretman, te se na taj način inhibira djelovanje adrenalnih androgena. Pacijentice sa KAH se uglavnom porađaju putem carskog reza, jer je vaginalni porod otežan ili nemoguć zbog hirurških korekcija dvosmislenih genitalija.
Reference:
- Medicinska fiziologija 11.izdanje, Arthur C.Guyton, M.D., John E. Hall, Ph.D., Savremena administracija, Beograd. 2008.
- Oxford handbook of endocrinology and diabetes 3 edition, John Wass, Katherine Owen.
- Marksove osnove medicinske biohemije; Michael Lieberman, PhD, Allan D.Marks, PhD, Colleen Smith, PhD; data status; Beograd. 2008.
- Clinical Endocrinology and Diabetes at Glance; Aled Rees, Miles Levy, Andrew Lansdown; WILEY Blackwell
- Patofiziologija 7.obnovljeno i dopunjeno izdanje, Stjepan Gamulin, Matko Marušič, Zdenko Kovač. Medicinska naklada. Zagred. 2011.
- “Kongenitalna adrenalna hiperplazija” Katja Dumić Kubat, Duje Braovac, Maja Vinković. Paediatr Croat. 2019; 63 (Supl 1): 81-86
- https://pubmed.ncbi.nlm.nih.gov/11359457/
- https://www.ncbi.nlm.nih.gov/books/NBK448098/
- https://pubmed.ncbi.nlm.nih.gov/28576284/
- https://pubmed.ncbi.nlm.nih.gov/25905188/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4544382/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5624825/