By Nerma Hodžić
•
February 16, 2025
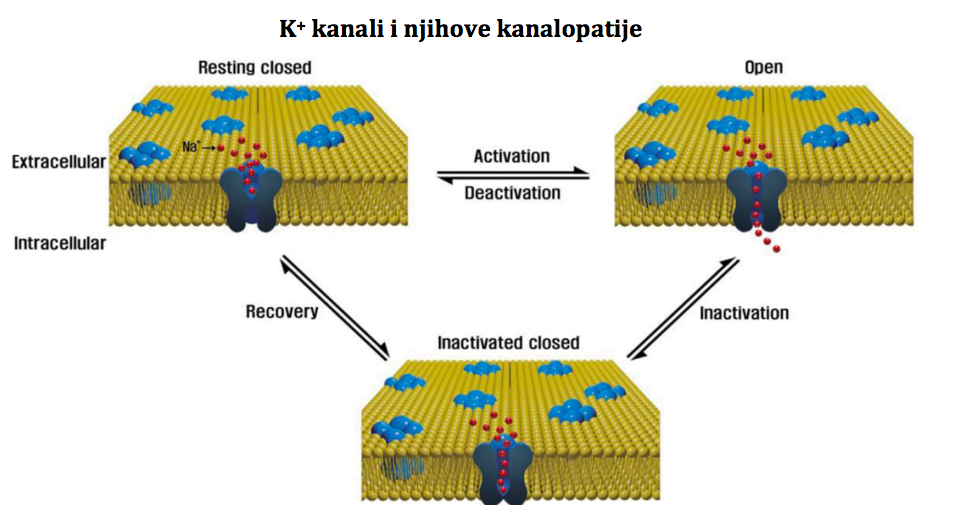
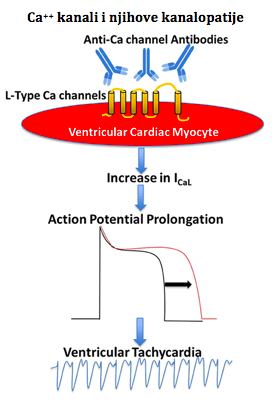
Moje ime je Nermina Hodžić. Majka sam dječaka Bakira Hodžića (10 godina), rođenog sa urođenom srčanom anomalijom hipoplazija lijeve strane srca - HLHS. Rođeni i živimo u Sarajevu. Dijagnozu smo saznali dan poslije rođenja. Rođen je kao beba u terminu, 4.150 gr težak, 56 cm dug, po rođenju odmah proplakao, apgar 9/10. Spontan porod, druga beba iz uredne trudnoće. U trudnoći niko nije mogao vidjeti anomaliju. Prvi dan nakon izlaska, beba počinje brzo da diše, orošen hladnim i ljepljivim znojem, blijed. Tu noć smo ga odveli doktoru gdje nam je rečeno da sam bebi dala previše hape i da je došlo do greške u ishrani. Bebi sam dala 20ml hape, jer sam se trudila uspostaviti dojenje. Tek kasnije sam shvatila da moja beba nije imala refleks sisanja i da nije ništa dojio jer nije imao snage. Vratili smo se kući, da bi nakon 4 sata opet odveli bebu na kliniku gdje su ga odmah primili i odveli na intezivnu njegu. Tu su ga reanimirali i stabilizovali ga. Došao je tada ljekar dr. Halimić Mirza i polako nam je pojasnio šta je bilo sa našom bebom. Sječam se da smo muž i ja sjedili u čekaonici dok doktor nije došao i samo smo molili Boga da bude živ. Kada je doktor došao, objasnio nam je sve o njegovoj dijagnozi i dao nam nadu da će sve biti dobro. Nisam znala i dan danas ne znam kako sam se osječala. Kao da se dešava nekome drugom. U jednom momentu mi je doktor crtao na papir normalno srce, na drugi papir mi je crtao srce moje bebe, kojeg nije bilo pola, i aorta koja je trebala biti velika i debela kod njega je bila mala i tanka (hipoplastična). Sjećam se da sam pitala: "K ako će on živjeti sa ovakvim srcem?" On nam je onda objasnio da djeca sa tim anomalijama prolaze kroz tri vrste operacija, i da na kraju imaju jedan kvalitetan i normalan život. Nisam to mogla u svojoj glavi sumirati, tada, jer mi je zvučalo nemoguće. Ne, nakon crteža i prognoza. Tada je dr. Halimić rekao da beba ostaje na intezivnoj i da će se prebaciti na Univerzitetsku kliniku AKH-a Beč, gdje će mu se uraditi prva pd tri operacije. 18 dana je bio na mašini, stabilan i dobar, 18-ti dan smo prebačeni u Austriju, a 20-ti dan je operisan. Operacija je trajala 12 sati, ali je prošlo sve u najboljem redu. Na intezivnoj njezi je bio 10 dana, i tu počinju problemi. Dobija sepsu, opet ide borba. Uspjeli su ga stabilizovati, i nakon 18 dana intenzivne njege prebacuju nas na odjel. Na odjelu opet problemi. Srce neće da pumpa, velike količine tekučine gubi, srčani ritam preko 300/min, jedna, druga, treća, četvrta transfuzija, 4 transfuzije albumina, 18 vrsta lijekova u perfuzomatima, doktori stalno ulaze i izlaze, razina kisika opada.... jedan korak naprijed napravimo, pet se vratimo unatrag. Sjećam se da mi je jednom tehničar ušao sa doktorima u sobu i rekao: " Mama, šta god bilo večeras sa vašim sinom, znajte da smo ovdje uz vas". Nisam ih registrovala šta mi pričaju, niti zašto, jer sve što sam ja radila je bilo gledanje u njega i u aparate iznad njega. Izdržali smo do jutra, preživio je, i došli su po njega da ga vode na intezivnu opet. Tada sam zamolila sestru da mi dopuste samo malo da ga držim, da ga pomilujem i pomirišem, da ga osjetim u rukama jer ga nisam uzela čitav mjesec. Namjestili su mi stolicu i dali mi ga. U momentu kada sam ga prinijela usnama, počela sam plakati nakon mjesec. Suze nisam pustila prije toga.. Ljuljala sam ga i ljubila, i u jednom momentu mom Bakiru su se vitalni parametri počeli vračati u normalu, sa 300-500 otkucaja na 100, saturacija sa 50 na 70%, on se zarumenio i lijepo počeo disati. Tada mi doktor dolazi nakon nekoh 4 ili 5 sati i kaže: "M ajka, nemojte ga ispuštati iz ruku, vi mu trebate ." I nisam... Nisam ga pustila nikako danima dok me nije opalio panični napad. Držala sam ga u rukama, i samo sam u jednom momentu osjetila kako nemam snage u lijevoj ruci, kako mi trne ruka i brada.. Pritisnem dugme za sestru, ona kada je došla uzima Bakira iz ruku i pita me šta je bilo. Ja nisam mogla da odgovorim, jer nisam imala sposobnost govora jer mi je sve utrnulo i jezik i lice i brada i ruke i noge. Nakon toga počinjem nekontrolisano da se tresem, čitavo tijelo, da su morali da me odnesu na krevet jer nisam bila u stanju sama hodati. Dali su mi nešto za smirenje i ja sam zaspala. Srce se smirilo, sve se malo ustabililo, ali tada Bakir dobija epi napade. Hitni CT pokaže promjene u vidu mikroishemijskih poremećaja. Na terapiji Keppra bio je dvije godine, i nikada se više nisu napadi ponovili. Druga operacija (Glenn)... Bakir ima 5,5 mjeseci, odlazimo na drugu op, koja protiče super, ali dolazi do komplikacija postoperativno. Hematom na Glennu, kojeg su hitno riješili dok je bio na intenzivnoj njezi. Poslije toga, na odjelu, hilotoraks lijevo, atelektaza lijevog plućnog krila, posebna hrana Monogen)… još mi kažu da ću ga morati hraniti na sondu. " Ma nema ništa od toga" , govorim im. Ja sam njega navikla da pije Monogen hapu jako neukusna i bljutava) na flašicu. Mjesec dana bolnice poslije Glenna. Treća operacija (Fontan)... E Fontan... Fontan je jedno čudo. Nisam vjerovala kada su mi ljekari govorili da ću ja znati kada je vrijeme za Fontan. Moj Bakir neposredno prije operacije nije mogao uz tri stepenice da se popne odmah se zamori, poplavi i sjedne. Sa 6 godina Baki dobija novu cirkulaciju. Operacija je trajala neka 3 sata, posoperativno je super sve prošlo. Samo što sam upozorila sestre na njega da se sam ekstubira. Nazvala sam tu noć da vidim kako je sestra mi je rekla da je super, i da se sam ekstubirao. Sutradan smo otišli muž i ja i kada smo ulazili na int.njegu čuli smo ga kako se svađa sa nekim, kad ono sestra koja ga je presvlačila, nije joj dao da ga obuče, htio je da ga ja prevučem. Bili smo u bolnici isto mjesec dana i na kraju otišli kući. Nedugo nakon toga počinje pandemija COVID-a. Moj Bakir dobija jaku infekciju gornjih disajnih puteva, ali nije izolovan COVID. 7 mjeseci poslije Fontan op., Bakir dobija plastični bronhitis. Nova borba, koja traje i dan danas. Plastični bronhitis je u biti poremečaj u limfnoj drenaži, usljed operacija i oštećenja limfnih puteva. Kod njega se manifestuje jakim opstrukcijama u disajnim putevima, tj. nakupljanjem cast-a. To je sadržaj gust i ljepljiv kao žvaka, a opet mekan kao spužva. Uglavnom, kako sam ja shvatila to je jedan vid propadanja Fontanove cirkulacije, a krajnji vid izlječenja te komplikacije je transplantacija srca. Njemu najbolje pomogne u tim napadima inhalacija Alteplaze (trombolitik)… koji odlijepi taj sadržaj i Ventolin sa 3%NaCl, da bi lakše iskašljao. Bakir je već godinu i po bio miran po pitanju tih opstrukcija, i prije mjesec dana mu se opet vratilo kako su počele oscilacije u vremnskim prilikama. Ja sam poslije Bakira rodila još jednu djevojčicu i to za vrijeme COVID-a. Uredna trudnoća, naravno obavila sam sve moguće pretrage vezane za razvoj, i hvala Bogu na kraju je sve bilo uredno. A kako je meni bilo? Iskreno, ne znam. Mislim da je mene lično kroz čitavo ovo putovanje koje je trajalo i traje i dan danas osnažila samo vjera u Boga. Bakira svi u porodici doživljavamo kao sasvim normalno dijete jer on to i jeste. Jako je aktivan, trenira fudbal, pametan je, ide u školu, 4 je razred, voli tjelesno najviše od svih predmeta, ima super društvo, voli se igrati i jako je duhovit. Nema nikakvih intelektualnih poteškoća hvala Bogu. Muž i ja smo shvatili da je adrenalin ono što ga pokreće. Samo neka je ekstremno. Sad ovo ljeto je naučio da roni na moru, i bez problema zaroni u dubinu do 3 m. i izroni, kada smo na moru ne izlazi iz vode, voli da skače sa stijena, sa skakaonica, sa pedalina... Ne mogu reći da me neke situacije ne izbace iz tračnica, ali to je tako u životu svakog roditelja koji ima dijete sa nekom anomalijom. Bilo fizičkom ili psihičkom. Moja sreća zaista je bila ta što sam ja kroz te operacije sa njim u bolnici imala i psihološku pomoć i podršku 2-3 puta sedmično u sklopu njegove rehabilitacije. Vjerujem i znam da mi je to održalo zdrav razum. Ja sam lično tip osobe koja je išla kroz zid glavom da bi svom djetetu pružila neophodnu pomoć i njegu I nema toga što me može zaustaviti kada je u pitanju traženje lijeka i pomoći, ne samo za njega, nego za sve troje. Šta bih poručila roditeljima? Poručila bih im sljedeće - Nema odustajanja!! Nije sve tako strašno. Jeste teško, bilo je teško, teško je i biće teško. Ali hvala Allahu na svemu i na svakom stanju. Od nečega ovako teškog uvijek ima teže. Koliko oni u svojim najslabijim momentima mogu nama da daju snage da guramo i borimo se nismo ni svjesni. Onog momenta kada se pomirite sa dijagnozom svog djeteta, i kada shvatite da ste tu gdje jeste, nema nazad, idete naprijed A naprijed vas čeka sunce, zelena livada, mir i smiraj. Naprijed vas čeka jedno biće prepuno ljubavi, sreće i milosti. Čeka vas vaša nagrada za koju ste se borili toliko teško, i toliko jako da ste mislili da nikad neće osvanuti dan kada će sve biti uredu. Dani i dani, mjeseci i godine su pred nama ako Bog da, a naše je samo da se zahvalimo dragom Bogu što je baš nas odabrao da budemo roditelji našim srčekovcima. Ne može to zaista svako. Kako moje dijete doživljava svoje stanje? To i jeste ustvari stanje, nije bolest. On sam za sebe kaže da je superheroj. Mislim da to sve govori o njemu. Nekad, ali nekad zaista osjetim da je pomalo anksiozan, pogotovo nakon što dobije ove optrukcije. On se toliko nasekira što ne može nekad da to iskašlje ili se nasekira ako počne kašljati hoće li moći zaspati, ali mi to uspijemo nekako prebroditi i preći preko toga. Zaista ja njim razgovaram kao sa odraslom osobom jer je jako pametan i inteligentan i razumije dosta toga . Motiviše me moja vjera. Motiviše me Bakir. Motiviše me svako novo otkriće i pronalazak nelog lijeka, riješenja, opcije itd.. Meni lično snagu daje moj Gospodar, jer bez Njega i Njegove podrške i pomoći ja ne bih ništa od ovoga mogla sama proći. Naravno, tu je muž i porodica, ali samo Gospodar je taj koji daje snagu i vjeru da će sve biti onako kako treba da bude. I kada se pomirimo sa činjenicom da je sve već propisano šta će biti i šta se dešava, lakše prihvatamo trenutno stanje jer znam da ima uvijek još nešto, ima neka nagrada koja nas sve čeka. Bakir je sad top. Super se osječa, super izgleda, dobrog zdravlja, bude tu i tamo nahlađen, bolestan, ali on je hvala Bogu dobro. Ko god ga vidi, a ne zna za njegovu dg, ljudi se zaprepaste kako on dobro izgleda kada saznaju. Jedina nam je sada briga i tražimo načine da nađemo bar nekog od stručnjaka vezano za plastični bronhitis. Ima načina, ima i ljekara, ali ke jako teško pronaći način da se dođe do njih. Ali, to je za neku drugu priču, ukoliko budete zainteresovani. Ja mislim da za ovo stanje ima načina u liječenju pored poznatih načina, ali trebamo samo nekoga ko bi nam pomogao da povežemo situacije i metode liječenja.